December 5, 2024 by University of Tokyo
Collected at: https://phys.org/news/2024-12-ray-method-crystal-multiphase-materials.html
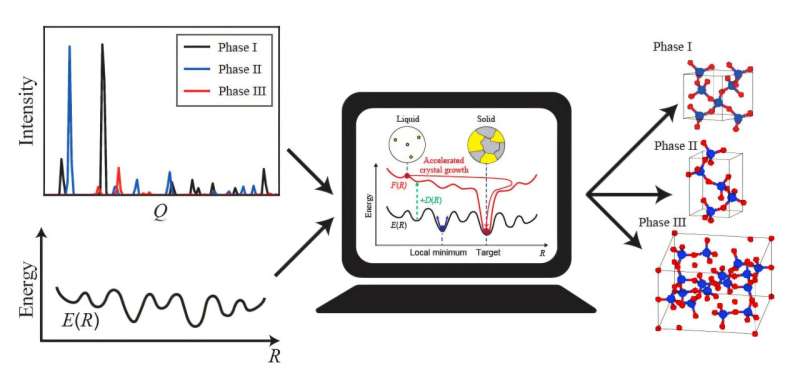
A joint research team led by Yuuki Kubo and Shiji Tsuneyuki of the University of Tokyo has developed a new computational method that can efficiently determine the crystal structures of multiphase materials, powders that contain more than one type of crystal structures. The method can predict the structure directly from powder X-ray diffraction patterns, the patterns of X-rays passing through crystals roughly the same size as instant coffee particles.
Unlike conventional methods, this approach does not require the use of “lattice constants” and can be applied to existing experimental data that could not be analyzed until now. Thus, the new method is a crucial asset for discovering new material phases and developing new materials. The findings are published in The Journal of Chemical Physics.
Many materials can have several crystal structures, “phases,” even in the same solid state. Determining the underlying crystal structures of materials is essential for understanding their properties and formulating strategies to develop new materials. However, conventional methods make calculations using the “lattice constant,” a property of the crystal being investigated.
In other words, they require knowledge about the crystal prior to determining its structure. This makes existing experimental data difficult to analyze when the lattice constant is not precisely known. Thus, there might be undiscovered crystal structures hidden in plain sight in already existing data.
“The crystal structures of the real world are extremely diverse. They are one of nature’s deepest mysteries,” says Kubo, the first author of the paper. “We thought that, in a way, we could get a glimpse of the depth of nature’s mysteries by developing our own method for determining unknown crystal structures.”
Conventional approaches employ various methods and are computationally costly. To reduce computational costs, the researchers set out to develop a method that can make predictions based on experimental data directly. They built their model based on molecular dynamics, simulating atomic motion by calculating the forces between atoms. Then, by incorporating experimental X-ray diffraction data, they enhanced the consistency between experimental data and simulations.
“We did not believe this method was promising,” says Kubo. “We were surprised when we ran the test calculations and the method performed far better than we had initially expected.”
The researchers confirmed the effectiveness of their method by applying it to widely researched materials. The method successfully reproduced the distinctive crystal structures of both carbon (graphite and diamond) and silicon dioxide (low-quartz, low-cristobalite, and coesite). It worked. However, Kubo is already thinking of the many possible next steps.
“We plan to apply this method to powder diffraction experimental data that have remained unutilized due to unsuccessful structural determination, aiming to discover new material phases. Furthermore, we aim to develop methods integrating experiments and simulations to determine not only crystal structures but also the structures of surfaces and interfaces.”
More information: Yuuki Kubo et al, Data-assimilated crystal growth simulation for multiple crystalline phases, The Journal of Chemical Physics (2024). DOI: 10.1063/5.0223390
Journal information: Journal of Chemical Physics
Leave a Reply