September 16, 2024 by University of Science and Technology of China
Collected at: https://phys.org/news/2024-09-strategy-simulating-nonadiabatic-dynamics-molecules.html
A research team has proposed a novel approach to accurately describe electron transfer mediated nonadiabatic dynamics of molecules at metal surfaces. Their works were published in Physical Review Letters.
Numerous experimental phenomena have demonstrated that non-adiabatic energy transfer is widespread in various interfacial processes. Therefore, studying non-adiabatic energy transfer is crucial for understanding interfacial processes such as chemical adsorption, electrochemistry, and plasmonic catalysis.
However, during the interaction between molecules and metal surfaces, molecular vibrations, rotations, and translations couple with surface phonons and electrons, leading to extremely complex energy transfer processes. Traditional models based on electronic friction have offered some insights, but they fall short in capturing the complex energy transfer observed in experimental studies.
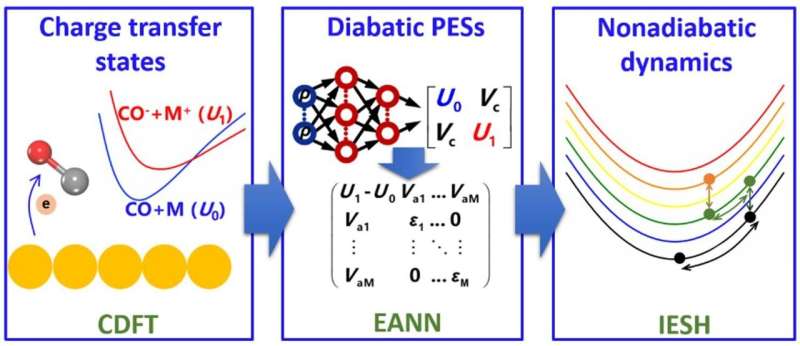
To tackle this problem, Prof. Jiang Bin from the University of Science and Technology of China of the Chinese Academy of Sciences and team developed a simulation strategy and applied it to the energy transfer dynamics of CO molecules scattering from AU(111) surfaces. The strategy starts by calculating the charge-transfer states of various configurations of CO molecules at the metal surfaces using constrained density functional theory (CDFT).
They then utilized an embedded atom neural network (EANN) to learn the CDFT energies and yield high-dimensional diabatic potential energy surfaces (PESs). Finally, they applied independent electron surface hopping (IESH) method to simulate the energy transfer process.
The results showed that the simulations closely matched the experimental data for the vibrational final state distribution of highly vibrationally excited CO (vi=17) after scattering. The vibrational relaxation probability, mean translational energy, and scattering angle distribution for low vibrationally excited CO (vi=2) were also accurately reproduced by the simulations.
Specifically, the simulation results also revealed different energy transfer pathways for different initial vibrational states. For high initial vibrational states, the molecular vibrational energy primarily transfers to surface electrons and molecular translation. In contrast, for low initial vibrational states, the molecular vibrational energy transfers exclusively to the surface electrons.
This study represents a significant advancement in understanding the energy transfer of a molecule-surface system. By providing a robust and accurate framework for modeling nonadiabatic dynamics, this strategy can be extended to studying other nonadiabatic dynamics at surfaces, potentially leading to future development in catalysis, materials science, and nanotechnology.
More information: Gang Meng et al, First-Principles Nonadiabatic Dynamics of Molecules at Metal Surfaces with Vibrationally Coupled Electron Transfer, Physical Review Letters (2024). DOI: 10.1103/PhysRevLett.133.036203
Journal information: Physical Review Letters
Leave a Reply